miRScore is a microRNA(miRNA) validation tool developed to analyze novel miRNAs annotated using small RNA-sequencing data. This tool can be used to determine whether MIRNA loci are of high confidence based on the criteria outlined in peer-reviewed publications. Users submit mature miRNA sequences, hairpin sequences, and sRNA-seq data to miRScore. miRScore will then score each MIRNA locus and output several files analyzing the submitted loci. The primary results can be found in the miRScore_Results.csv file, which contains positional and read information, as well as a pass/fail result for each MIRNA locus.
Determining what is a ‘miRNA’
What constitutes a miRNA is based on several structural and quantitative characteristics. For full details on these criteria, please see miRScore: a rapid and precise microRNA validation tool. Here is a short summary of the criteria outlined in the manuscript:
microRNA Criteria
Less than five mismatches in miRNA duplex
There must not be more than three nucleotides in asymmetric bulges
Two nucleotide 3’ overhang of miRNA duplex
No secondary stems or large loops in miRNA duplex
miRNA must be between 20-24 nucleotides for plants or 20-26 for animals
Confirmation of the miRNA and miRNA* by small RNA-sequencing
At least 75% of reads that map to hairpin must come from miRNA duplex
At least ten exact reads must map to the miRNA duplex within a single library
miRScore verifies MIRNA loci using these conditions. miRScore utilizes a read floor which requires at least 10 exact miRNA/miRNA* reads map to the miRNA duplex in at least one library.
Installation
miRScore can be installed through conda package manager from the bioconda channel, or through manual set up.
Installing using conda - Mac/Linux environment
First install conda. Once installed, you can create and activate an environment for miRScore using the following commands:
Download miRScore script from the GitHub repo. miRScore can be run using python by placing the script in your working directory. You can make the script executable using chmod +x miRScore and copying it to your environment’s PATH.
FASTA file containing mature and star sequences of miRNAs
hairpin
FASTA file containing hairpin precursor sequences of mirnas
fastq
List of small RNA-seq libraries in FASTQ or FASTA format
mm
Allow up to 1 mismatch in sRNA-seq reads
n
Specify a name to be added at the beginning of output files
threads
Specify number of threads to use during trimming step
autotrim
Trim fastq files using cutadapt
trimkey
miRNA sequence for detecting adapter sequences for trimming
kingdom
Specify either ‘plant’ or ‘animal’
out
Specify output directory. Defaults to ‘miRScore_output/‘
rescue
Reevaluate failed miRNAs and reannotate locus with alternative miRNA duplex that meets all criteria
Bowtie
Bowtie has been configured to run the following options.
a, Report all valid alignments
v, Allow 0 or 1 mismatch depending on option ‘mm’
-no-unal, No unaligned reads reported
-norc, No reverse reads reported, due to reads being aligned to a single stranded hairpin and not a genome
Rather than mapping to a genome, miRScore uses Bowtie to map each read to the MIRNA hairpin.
Required
miRScore requires two FASTA files and small RNA-seq libraries in FASTQ format in order to run.
-mature maturefile: FASTA file containing mature miRNA sequences of proposed novel miRNAs for scoring.
-hairpin hairpinfile: FASTA file containing the hairpin sequences in which the mature miRNAs can be found.
-fastq fastqfiles: Small RNA-seq libraries in FASTQ or FASTA format (can be compressed). If libraries are untrimmed, you must use option ‘-autotrim’ to trim libraries. You may supply a key for trimming using option ‘-trimkey’. Default trimkey is ATH-miR166a for plants and HSA-let-7a for animals. Ideally, all FASTQ files should be unique individual libraries. If merged libraries are provided, be sure not to include any individual libraries present in the merged file, as this may cause issues with read counting. It is recommended to avoid the use of merged libraries when possible to ensure highest confidence in miRNA validation. See FAQ for more details.
Please note: the sequence identifier of each miRNA (i.e. ATH-miR173) must match the sequence identifier of the corresponding hairpin precursors in the hairpin file (i.e. ATH-MIR173)! This is not case sensitive.
Example
For this example, miRScore was tested on a set of 15 Arabidopsis thalianaMIRNA loci downloaded from miRBase and small RNA-sequencing data downloaded from Lunardon et al., (2020).
Download the two FASTA files provided in TestData folder: ath_miRBase_miRNAs.fa and ath_miRBase_precursors.fa. Place these in your working directory.
Create a directory for small RNA-seq libraries and retrieve FASTQ files.
mkdir fastqs
cd fastqs
fasterq-dump SRR3222443 SRR3222444 SRR3222445
cd ..
A common use case for miRScore may include evaluating known miRNAs from databases such as miRBase or MirGeneDB using user-generated sRNA-seq libraries. Both databases have inconsistencies and idiosyncrasies in their microRNA and hairpin naming schemes that prevent input of “raw” files into miRScore. Here are two examples of how to prepare data from miRBase and MirGeneDB respectively.
miRBase
Example : Arabidopsis thaliana
1. Download the ‘mature.fa’ and ‘hairpin.fa’ file from miRBase download page.
At this stage, if you run miRScore it will inform you of the following:
Error! The following entries in the MIRNA hairpin file ‘ath_hp.fa’ have no mature sequences that match their identifiers in file ‘ath_mat.fa’. [‘ath-MIR161’, ‘ath-MIR779’, ‘ath-MIR780’, ‘ath-MIR869’, ‘ath-MIR1886’]
Please check all hairpins in the hairpin FASTA file have miRNAs in the mature FASTA file with the same name.
If you have multiple miRNAs assigned to a single locus (i.e. osa-miR159a.1/osa-miR159a.2 to osa-MIR159a)
please run ‘hairpinHelper’ then rerun miRScore with the’miRScore_adjusted_hairpins.fa’ file as the hairpin FASTA input.
miRScore_Results.csv: A csv file containing the results for all proposed miRNAs.
reads.csv: A csv file containing read counts for miR, miR*, and total reads mapped to hairpin in each submitted library.
alt_miRScore_Results.csv: A csv file containing the results for alternative miRNA suggestions on a failed miRNA hairpin.
alt_reads.csv: A csv file containing read counts for alternative miR, miR*, and total reads mapped to hairpin in each submitted library
RNAplots: Directory containing PDF images for each miRNA secondary structure including miR(red)/miR*(green) annotation.
strucVis: Directory containing PDF images depicting miRNA secondary structure and small RNA read depth for each MIRNA locus.
miRScore_Results.csv
This file contains the results for all candidate miRNAs submitted to miRScore. Do NOT edit this file if submitting to miRBase. miRBase submission requires specific formatting, specified by miRScore output. Editing this file may cause issues in submission.
name - MIRNA name provided by user.
mSeq - mature miRNA sequence.
mLen - mature miRNA length.
mStart - Start position of mature miRNA on hairpin precursor.
mStop - Stop position of mature miRNA on hairpin precursor.
msSeq- miRNA star sequence.
msLen - miRNA star length.
msStart - Start position of miRNA star on hairpin precursor.
msStop - Stop position of miRNA star on hairpin precursor.
precSeq - Hairpin precursor sequence.
precLen - Length of hairpin precursor.
result - Pass/Fail result of miRScore for miRNA.
flags - Criteria that was not met by the MIRNA locus.
mismatch - Number of mismatches in miRNA duplex.
mReads - Sum of reads that map to mature miRNA position +/- 1 position in all libraries which meet criteria. See FAQ1 for details on read counting.
msReads - Sum of reads that map to miRNA star position +/- 1 position. See FAQ1 for details on read counting.
totReads - Total reads that map to the hairpin precursor in all libraries which meet criteria. See FAQ1 for details on read counting. Please note this does not always equal mReads + msReads. This is ALL reads that map to the hairpin.
numLibraries - Number of libraries that reads were report from for mReads, msReads, and totReads.
precision - Measure of precision of how many reads map to the miRNA duplex compared to other positions in the precursor. Found by totaling the number of reads for miR and miR*, then dividing by the sum of all reads mapped to the hairpin precursor. (mReads + msReads) / totReads
Flags
Flags are reported to help the user determine what criteria a MIRNA locus did not meet. Most flags reported lead to a hard fail (i.e. ‘Precision less than 75%’, ‘Less than 10 reads’). Two flags (‘23/24 nt’ and ‘Precursor > 300 nt’) do not cause a MIRNA locus to fail, and instead are reported to indicate to the user that the miRNA or the MIRNA hairpin have longer lengths and should be evaluted carefully.
List of potential flags
Flag
Explanation
Result if flag is present
More than 5 mismatches in duplex
More than 5 base pairs are mismatched in the miRNA duplex
fail(plants) / warning (animals)
More than 7 mismatches in the duplex
More than 7 base pairs are mismatched in the miRNA duplex
Fail
23/24 nt miRNA
The miRNA/miRNA* duplex is 23 or 24 nucleotides in length
Warning
Asymmetric bulge greater than 3
There is an asymmetric bulge greater than 3 base pairs in the miRNA duplex
Fail
Hairpin is less than 50 nucleotides
The user-provided hairpin sequence is less than 50 nucleotides in length
Fail
miRNA multimaps to hairpin
The miRNA or miRNA* provided by the user indexed to multiple locations on the hairpin
Fail
Less than 10 reads in a single library
Less than 10 combined miRNA/miRNA* reads in a single library were detected. Does not meet the read floor.
Fail
No mature or star reads detected
No reads were detected for the miRNA or miRNA* in a single library
Fail
Precision less than 75%
The precision (miRNA reads + miRNA* reads/total reads mapped to hairpin) did not reach 75% in a single library
Fail
No 2nt 3’ overhang
The user-provided miRNA/miRNA* sequences did not form a duplex with a 2nt 3’ overhang
Fail
Hairpin structure invalid
The hairpin secondary structure did not allow indexing of miRNA duplex. This may be due to large bulge or secondary stem loop.
Fail
Star length not met
The miRNA star sequence is less than or greater than allowed by criteria
Fail
Mature miRNA length not met
The mature miRNA length is less than or greater than allowed by criteria
Fail
Precursor > 300 nt
The hairpin sequence is larger than 300 nucleotides
Warning (plants)
Precursor > 200 nt
The hairpin sequence is larger than 200 nucleotides
Warning (animals)
reads.csv
A csv file containing the reads reported in each library of the miRNA, miRNA*, and total reads mapped to each hairpin. Also includes a Pass/Fail result and flag column. A ‘Pass’ indicates the MIRNA locus that meets the read floor (greater than 10 reads map to the miRNA duplex), has at least one read mapped to both miR and miR*, and has a precision greater than 75% in the reported library. A ‘Fail’ indcates one of these criteria are not met.
alt_mirna_results.csv
This is an optional result when the option ‘-rescue’ is used. When a miRNA ‘fails’ miRScore, meaning one or more of the criteria are not met, that locus will be reavaluated based on sequencing data. This revaluation is done using the reads mapped to the hairpin associated with the failed MIRNA locus. miRScore will use samtools to select the most abundant read in the alignments/merged.bam file with a sequence between 20-24 nt that mapped to the failed MIRNA hairpin. miRScore will then score that sequence as an alternative miRNA. Any of these alternative miRNAs that pass will be reported in alt_miRScore_Results.csv. These alternative miRNAs are not intended to be final confirmation of a MIRNA locus, and should be evaluated with care.
alt_reads.csv
A csv file containing the reads reported in each library of the alternative miRNA, alternative miRNA*, and total reads mapped to each hairpin. Also includes a Pass/Fail result. A ‘Pass’ indicates the alternative miRNA meets the read floor and has a precision greater than 75% in that library. A ‘Fail’ indcates one of these criteria are not met.
RNAplots
Directory containing PDF files of MIRNA secondary structure. This structure is predicted using Vienna RNAfold. For more information on RNA predicted structures, please visit ViennaRNA page.
Each file contains the hairpin sequence for a miRNA, as well as annotation for the miRNA and predicted miRNA* sequences. The miRNA is outlined in red, while the miRNA* is outlined in green.
strucvis_plots
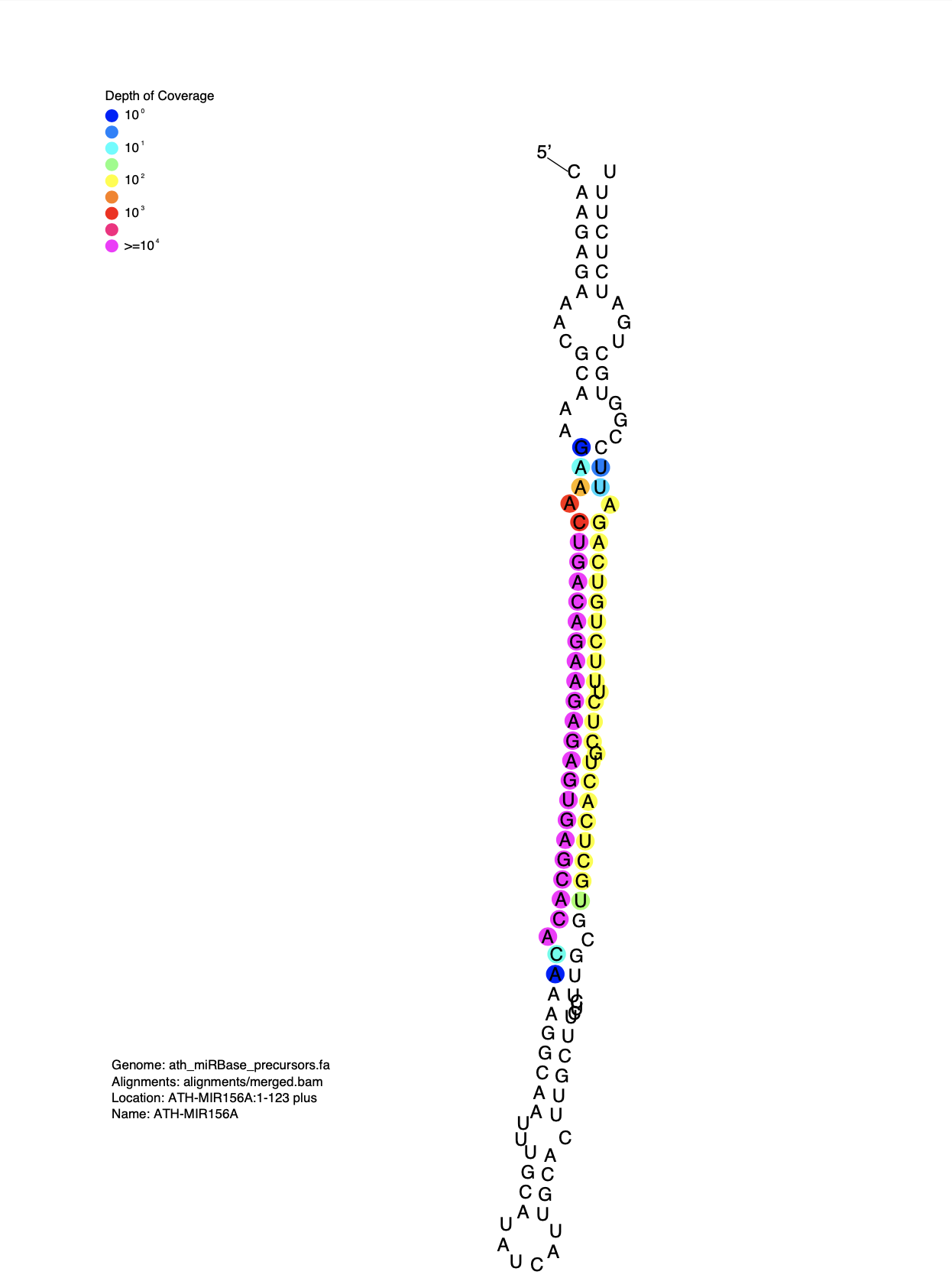
Directory containing PDF images generated by strucVis of MIRNA secondary structure including small RNA read depth of coverage.
hairpinHelper
hairpinHelper was created to address an issue with some datasets where there are multiple miRNAs in the mature FASTA file correspond to a single hairpin sequence in the hairpin FASTA file. This would occur when multiple miRNAs come from a single locus.
miRScore requires that each hairpin have only one corresponding miRNA. This is because the way miRScore handles MIRNA names in the programming. To help users solve this issue, we’ve included this script to quickly resolve the instance where two or more miRNAs (i.e. osa-miR159a.1 and osa-miR159a.2) have only a single locus present in the hairpin file (osa-MIR159a).
This script will find any miRNAs that have the same name as the hairpin and create a duplicate hairpin locus (same sequence) but with the name of the miRNA from the mature file.
The result is a FASTA file called ‘miRScore_adjusted_hairpins.fa’. After generating this file, rerun miRScore using this FASTA file as the input for the ‘-hairpin’ option.
FASTA file containing mature and star sequences of miRNAs
hairpin
FASTA file containing hairpin precursor sequences of mirnas
FAQ
1. How does miRScore handle reporting reads?
miRScore reports read counts for the miRNA duplex, totReads, and precision based on which sRNA-seq libraries pass or fail. If a user submits 10 libraries, and all the expression criteria are met in only 2, mreads, msReads, totReads, and precision will reflect only those 2 libraries, while the remaining 8 will not be included. Read counts for each locus in each of the 10 libraries can still be found in ‘reads.csv’ file. If either expression criterion is not met in any individual library, the miRNA will Fail and miRScore will report these values for all 10 libraries.
2. Why do some of my miRNAs have ‘NA’s in certain columns?
MIRNA secondary structure plays a big role in determining many of the miRNA criteria. If the miRNA or miRNA* sequences index to the stem loop or large secondary loop, the miRNA duplex can be impossible to determine. Without this duplex, it’s not possible to score a MIRNA locus. In this instance, miRScore will report only the user-provided information and report ‘NA’s in all other columns.
3. Why is my miRNA failing due to no reads detected, but I see reads reported in both ‘mReads’ and ‘msReads’?
miRscore counts reads in individual sRNA-seq libraries. In order for a MIRNA locus to pass, there must be reads mapping to both the miRNA and miRNA* positions within a single library. This is to prevent false positive validation of miRNAs with a single read in dozens of libraries that may occur if a merged library was used.
miRScore reports three read count values in the results file: mReads, msReads, and totReads. mReads and msReads are counted using the reported locations of the miRNA/miRNA* on the hairpin +/- 1 position to account for variance. If no reads are detected at one or both of these positions, miRScore will flag that locus as “No mature or star reads detected”. In some cases, there are reads mapping to other regions of the hairpin, but not to the miRNA duplex. If this happens, you may see mReads or msReads reported as 0 while totReads is greater than 0. This is because totReads is the total number of reads that map to the hairpin, not just the miRNA duplex.
In other cases, you may see “No mature or star reads detected”, but mReads and msReads is reported to be greater than 0. This will only occur if you have multiple libraries submitted and miRNA/miRNA* reads are not detected within a single library for ANY library. Meaning, each library contains only miRNA OR miRNA* reads, but not both. If all libraries fail expression criteira, miRScore will report combined read counts for all of the failed libraries. To inspect the read counts in individual libraries, please see the ‘reads.csv’ file output by miRScore.
4. Why is the use of merged libraries not recommended?
As stated in FAQ 3, this is to prevent false positive validation of a MIRNA locus with a single read in dozens of libraries that may occur if a merged library was used.
The use of merged libraries is not suggested because one of the primary criteria miRScore uses as validation of miRNAs is ensuring both the miRNA and miRNA* is expressed within a single library. This provides a level of confidence that the miRNA is expressed at detectable levels within the sample, and that the reads are in fact miRNA reads with the characteristic two-stack mapping pattern. In some instances, miRNAs annotated by existing sRNA annotation tools only pass when the merged library is used, as it is common practice to use merged libraries to annotate miRNAs. Please be assured that miRScore will still handle merged libraries accurately, but the miRNA and miRNA* abundance reported in the results will reflect the overall abundance and not that of individual libraries. Please see FAQ 1 on how reads are reported when multiple libraries are submitted.
5. What is the rescue option and how is this useful?
The rescue option allows you to further investigate loci that failed miRScore with your provided miRNA/miRNA* sequences. This process identifies the most abundant read of the appropriate length that maps to the hairpin you provided for the locus and assigns this as the mature miRNA. miRScore will then reanalyze the locus with the newly assign mature miRNA and score it accordingly. If all miRNA criteria are met with this ‘rescued’ locus, the results are reported to ‘alt_mirna_results.csv’. Any potential “rescued” loci that emerge from this optional pipeline should be scrutinized manually before final annotation and submission to a miRNA registry.
6. Why is the miRNA sequence I submitted in the star position in the results file?
A mature miRNA is one which is taken up by the RNA-induced silencing complex (RISC) and guides post-transcriptional gene silencing. The mature miRNA strand is generally more abundant than the miRNA* because following RISC formation, the miRNA* is often rapidly degraded and therefore not detectable through sequencing. Therefore, when annotating miRNAs that lack evidence for which strand is functional, a common practice is to evaluate abundance of each strand of the miRNA duplex to determine which is likely the mature miRNA. The final step of miRScore is to reevalute each submitted locus and determine which strand of the final reported duplex is more abundant, then reassign that sequence as the mature miRNA. All identifiers/miRNA names are retained in this process.


Table of Contents
Overview
miRScore is a microRNA(miRNA) validation tool developed to analyze novel miRNAs annotated using small RNA-sequencing data. This tool can be used to determine whether MIRNA loci are of high confidence based on the criteria outlined in peer-reviewed publications. Users submit mature miRNA sequences, hairpin sequences, and sRNA-seq data to miRScore. miRScore will then score each MIRNA locus and output several files analyzing the submitted loci. The primary results can be found in the miRScore_Results.csv file, which contains positional and read information, as well as a pass/fail result for each MIRNA locus.
Determining what is a ‘miRNA’
What constitutes a miRNA is based on several structural and quantitative characteristics. For full details on these criteria, please see miRScore: a rapid and precise microRNA validation tool. Here is a short summary of the criteria outlined in the manuscript:
miRScore verifies MIRNA loci using these conditions. miRScore utilizes a read floor which requires at least 10 exact miRNA/miRNA* reads map to the miRNA duplex in at least one library.
Installation
miRScore can be installed through
condapackage manager from the bioconda channel, or through manual set up.Installing using conda - Mac/Linux environment
First install conda. Once installed, you can create and activate an environment for miRScore using the following commands:
Installing manually
Dependencies
The following packages are required to run miRScore V0.3.0. Create an environment with the following packages:
python>=3.6 https://www.python.orgbiopythonhttps://biopython.org/bowtie>=1.3.1 https://bowtie-bio.sourceforge.net/index.shtmlsamtools>=1.16 https://www.htslib.org/viennarna>=2.5.1 https://www.tbi.univie.ac.at/RNA/documentation.htmlpysam>=0.21.0 https://pysam.readthedocs.io/en/latest/api.htmltqdm>=4.65 https://pypi.org/project/tqdm/pandas>=2.0.0 https://pandas.pydata.org/cutadapt>=4.8 https://cutadapt.readthedocs.io/en/stable/guide.htmlstrucVis>=0.8 https://anaconda.org/bioconda/strucvisDownload miRScore script from the GitHub repo. miRScore can be run using python by placing the script in your working directory. You can make the script executable using
chmod +x miRScoreand copying it to your environment’s PATH.Usage
miRScore
Options
Bowtie
Bowtie has been configured to run the following options.
Rather than mapping to a genome, miRScore uses Bowtie to map each read to the MIRNA hairpin.
Required
miRScore requires two FASTA files and small RNA-seq libraries in FASTQ format in order to run.
-mature maturefile: FASTA file containing mature miRNA sequences of proposed novel miRNAs for scoring.-hairpin hairpinfile: FASTA file containing the hairpin sequences in which the mature miRNAs can be found.-fastq fastqfiles: Small RNA-seq libraries in FASTQ or FASTA format (can be compressed). If libraries are untrimmed, you must use option ‘-autotrim’ to trim libraries. You may supply a key for trimming using option ‘-trimkey’. Default trimkey is ATH-miR166a for plants and HSA-let-7a for animals. Ideally, all FASTQ files should be unique individual libraries. If merged libraries are provided, be sure not to include any individual libraries present in the merged file, as this may cause issues with read counting. It is recommended to avoid the use of merged libraries when possible to ensure highest confidence in miRNA validation. See FAQ for more details.Please note: the sequence identifier of each miRNA (i.e. ATH-miR173) must match the sequence identifier of the corresponding hairpin precursors in the hairpin file (i.e. ATH-MIR173)! This is not case sensitive.
Example
For this example, miRScore was tested on a set of 15 Arabidopsis thaliana MIRNA loci downloaded from miRBase and small RNA-sequencing data downloaded from Lunardon et al., (2020).
Download the two FASTA files provided in TestData folder:
ath_miRBase_miRNAs.faandath_miRBase_precursors.fa. Place these in your working directory.Create a directory for small RNA-seq libraries and retrieve FASTQ files.
Preparing database files
A common use case for miRScore may include evaluating known miRNAs from databases such as miRBase or MirGeneDB using user-generated sRNA-seq libraries. Both databases have inconsistencies and idiosyncrasies in their microRNA and hairpin naming schemes that prevent input of “raw” files into miRScore. Here are two examples of how to prepare data from miRBase and MirGeneDB respectively.miRBase
Example : Arabidopsis thaliana
1. Download the ‘mature.fa’ and ‘hairpin.fa’ file from miRBase download page.
2. Prepare hairpin file by converting to single line FASTA
3. Parse out ath entries and adjust names to be more concise. The mature miRNA and hairpin names must match!
4. Download fastq files
At this stage, if you run miRScore it will inform you of the following:
Error! The following entries in the MIRNA hairpin file ‘ath_hp.fa’ have no mature sequences that match their identifiers in file ‘ath_mat.fa’. [‘ath-MIR161’, ‘ath-MIR779’, ‘ath-MIR780’, ‘ath-MIR869’, ‘ath-MIR1886’]
Please check all hairpins in the hairpin FASTA file have miRNAs in the mature FASTA file with the same name. If you have multiple miRNAs assigned to a single locus (i.e. osa-miR159a.1/osa-miR159a.2 to osa-MIR159a) please run ‘hairpinHelper’ then rerun miRScore with the’miRScore_adjusted_hairpins.fa’ file as the hairpin FASTA input.
So you need to run hairpinHelper like so:
Alternatively, you could just remove those entries from the hairpin file.
5. Run miRScore using adjusted hairpins
MirGeneDB
Example : Homo sapiens
1. Download precursors w/ flank and mature sequences
2. Adjust precursors to not have ‘_pri’ and miRNAs need to have ‘-3p’ not “_3p” in order for miRScore to recognize.
3. Download fastq files
4. Run miRScore
Output
miRScore has six outputs:
miRScore_Results.csv: A csv file containing the results for all proposed miRNAs.reads.csv: A csv file containing read counts for miR, miR*, and total reads mapped to hairpin in each submitted library.alt_miRScore_Results.csv: A csv file containing the results for alternative miRNA suggestions on a failed miRNA hairpin.alt_reads.csv: A csv file containing read counts for alternative miR, miR*, and total reads mapped to hairpin in each submitted libraryRNAplots: Directory containing PDF images for each miRNA secondary structure including miR(red)/miR*(green) annotation.strucVis: Directory containing PDF images depicting miRNA secondary structure and small RNA read depth for each MIRNA locus.miRScore_Results.csv
This file contains the results for all candidate miRNAs submitted to miRScore. Do NOT edit this file if submitting to miRBase. miRBase submission requires specific formatting, specified by miRScore output. Editing this file may cause issues in submission.
name- MIRNA name provided by user.mSeq- mature miRNA sequence.mLen- mature miRNA length.mStart- Start position of mature miRNA on hairpin precursor.mStop- Stop position of mature miRNA on hairpin precursor.msSeq- miRNA star sequence.msLen- miRNA star length.msStart- Start position of miRNA star on hairpin precursor.msStop- Stop position of miRNA star on hairpin precursor.precSeq- Hairpin precursor sequence.precLen- Length of hairpin precursor.result- Pass/Fail result of miRScore for miRNA.flags- Criteria that was not met by the MIRNA locus.mismatch- Number of mismatches in miRNA duplex.mReads- Sum of reads that map to mature miRNA position +/- 1 position in all libraries which meet criteria. See FAQ1 for details on read counting.msReads- Sum of reads that map to miRNA star position +/- 1 position. See FAQ1 for details on read counting.totReads- Total reads that map to the hairpin precursor in all libraries which meet criteria. See FAQ1 for details on read counting. Please note this does not always equal mReads + msReads. This is ALL reads that map to the hairpin.numLibraries- Number of libraries that reads were report from for mReads, msReads, and totReads.precision- Measure of precision of how many reads map to the miRNA duplex compared to other positions in the precursor. Found by totaling the number of reads for miR and miR*, then dividing by the sum of all reads mapped to the hairpin precursor. (mReads + msReads) / totReadsFlags
Flags are reported to help the user determine what criteria a MIRNA locus did not meet. Most flags reported lead to a hard fail (i.e. ‘Precision less than 75%’, ‘Less than 10 reads’). Two flags (‘23/24 nt’ and ‘Precursor > 300 nt’) do not cause a MIRNA locus to fail, and instead are reported to indicate to the user that the miRNA or the MIRNA hairpin have longer lengths and should be evaluted carefully.
List of potential flags
reads.csv
A csv file containing the reads reported in each library of the miRNA, miRNA*, and total reads mapped to each hairpin. Also includes a Pass/Fail result and flag column. A ‘Pass’ indicates the MIRNA locus that meets the read floor (greater than 10 reads map to the miRNA duplex), has at least one read mapped to both miR and miR*, and has a precision greater than 75% in the reported library. A ‘Fail’ indcates one of these criteria are not met.
alt_mirna_results.csv
This is an optional result when the option ‘-rescue’ is used. When a miRNA ‘fails’ miRScore, meaning one or more of the criteria are not met, that locus will be reavaluated based on sequencing data. This revaluation is done using the reads mapped to the hairpin associated with the failed MIRNA locus. miRScore will use samtools to select the most abundant read in the alignments/merged.bam file with a sequence between 20-24 nt that mapped to the failed MIRNA hairpin. miRScore will then score that sequence as an alternative miRNA. Any of these alternative miRNAs that pass will be reported in alt_miRScore_Results.csv. These alternative miRNAs are not intended to be final confirmation of a MIRNA locus, and should be evaluated with care.
alt_reads.csv
A csv file containing the reads reported in each library of the alternative miRNA, alternative miRNA*, and total reads mapped to each hairpin. Also includes a Pass/Fail result. A ‘Pass’ indicates the alternative miRNA meets the read floor and has a precision greater than 75% in that library. A ‘Fail’ indcates one of these criteria are not met.
RNAplots
Directory containing PDF files of MIRNA secondary structure. This structure is predicted using Vienna RNAfold. For more information on RNA predicted structures, please visit ViennaRNA page.
Each file contains the hairpin sequence for a miRNA, as well as annotation for the miRNA and predicted miRNA* sequences. The miRNA is outlined in red, while the miRNA* is outlined in green.

strucvis_plots
Directory containing PDF images generated by strucVis of MIRNA secondary structure including small RNA read depth of coverage.
hairpinHelper
hairpinHelper was created to address an issue with some datasets where there are multiple miRNAs in the mature FASTA file correspond to a single hairpin sequence in the hairpin FASTA file. This would occur when multiple miRNAs come from a single locus.
miRScore requires that each hairpin have only one corresponding miRNA. This is because the way miRScore handles MIRNA names in the programming. To help users solve this issue, we’ve included this script to quickly resolve the instance where two or more miRNAs (i.e. osa-miR159a.1 and osa-miR159a.2) have only a single locus present in the hairpin file (osa-MIR159a).
This script will find any miRNAs that have the same name as the hairpin and create a duplicate hairpin locus (same sequence) but with the name of the miRNA from the mature file.
The result is a FASTA file called ‘miRScore_adjusted_hairpins.fa’. After generating this file, rerun miRScore using this FASTA file as the input for the ‘-hairpin’ option.
Usage
Options
FAQ
1. How does miRScore handle reporting reads?
miRScore reports read counts for the miRNA duplex, totReads, and precision based on which sRNA-seq libraries pass or fail. If a user submits 10 libraries, and all the expression criteria are met in only 2,
mreads,msReads,totReads, andprecisionwill reflect only those 2 libraries, while the remaining 8 will not be included. Read counts for each locus in each of the 10 libraries can still be found in ‘reads.csv’ file. If either expression criterion is not met in any individual library, the miRNA will Fail and miRScore will report these values for all 10 libraries.2. Why do some of my miRNAs have ‘NA’s in certain columns?
MIRNA secondary structure plays a big role in determining many of the miRNA criteria. If the miRNA or miRNA* sequences index to the stem loop or large secondary loop, the miRNA duplex can be impossible to determine. Without this duplex, it’s not possible to score a MIRNA locus. In this instance, miRScore will report only the user-provided information and report ‘NA’s in all other columns.
3. Why is my miRNA failing due to no reads detected, but I see reads reported in both ‘mReads’ and ‘msReads’?
miRscore counts reads in individual sRNA-seq libraries. In order for a MIRNA locus to pass, there must be reads mapping to both the miRNA and miRNA* positions within a single library. This is to prevent false positive validation of miRNAs with a single read in dozens of libraries that may occur if a merged library was used.
miRScore reports three read count values in the results file:
mReads,msReads, andtotReads.mReadsandmsReadsare counted using the reported locations of the miRNA/miRNA* on the hairpin +/- 1 position to account for variance. If no reads are detected at one or both of these positions, miRScore will flag that locus as “No mature or star reads detected”. In some cases, there are reads mapping to other regions of the hairpin, but not to the miRNA duplex. If this happens, you may seemReadsormsReadsreported as 0 whiletotReadsis greater than 0. This is becausetotReadsis the total number of reads that map to the hairpin, not just the miRNA duplex.In other cases, you may see “No mature or star reads detected”, but
mReadsandmsReadsis reported to be greater than 0. This will only occur if you have multiple libraries submitted and miRNA/miRNA* reads are not detected within a single library for ANY library. Meaning, each library contains only miRNA OR miRNA* reads, but not both. If all libraries fail expression criteira, miRScore will report combined read counts for all of the failed libraries. To inspect the read counts in individual libraries, please see the ‘reads.csv’ file output by miRScore.4. Why is the use of merged libraries not recommended?
As stated in FAQ 3, this is to prevent false positive validation of a MIRNA locus with a single read in dozens of libraries that may occur if a merged library was used.
The use of merged libraries is not suggested because one of the primary criteria miRScore uses as validation of miRNAs is ensuring both the miRNA and miRNA* is expressed within a single library. This provides a level of confidence that the miRNA is expressed at detectable levels within the sample, and that the reads are in fact miRNA reads with the characteristic two-stack mapping pattern. In some instances, miRNAs annotated by existing sRNA annotation tools only pass when the merged library is used, as it is common practice to use merged libraries to annotate miRNAs. Please be assured that miRScore will still handle merged libraries accurately, but the miRNA and miRNA* abundance reported in the results will reflect the overall abundance and not that of individual libraries. Please see FAQ 1 on how reads are reported when multiple libraries are submitted.
5. What is the rescue option and how is this useful?
The rescue option allows you to further investigate loci that failed miRScore with your provided miRNA/miRNA* sequences. This process identifies the most abundant read of the appropriate length that maps to the hairpin you provided for the locus and assigns this as the mature miRNA. miRScore will then reanalyze the locus with the newly assign mature miRNA and score it accordingly. If all miRNA criteria are met with this ‘rescued’ locus, the results are reported to ‘alt_mirna_results.csv’. Any potential “rescued” loci that emerge from this optional pipeline should be scrutinized manually before final annotation and submission to a miRNA registry.
6. Why is the miRNA sequence I submitted in the star position in the results file?
A mature miRNA is one which is taken up by the RNA-induced silencing complex (RISC) and guides post-transcriptional gene silencing. The mature miRNA strand is generally more abundant than the miRNA* because following RISC formation, the miRNA* is often rapidly degraded and therefore not detectable through sequencing. Therefore, when annotating miRNAs that lack evidence for which strand is functional, a common practice is to evaluate abundance of each strand of the miRNA duplex to determine which is likely the mature miRNA. The final step of miRScore is to reevalute each submitted locus and determine which strand of the final reported duplex is more abundant, then reassign that sequence as the mature miRNA. All identifiers/miRNA names are retained in this process.