
目录

- Rupdated mutational data processing functions1年前
- instInitial commit5年前
- manupdated mutational data processing functions1年前
- testsInitial commit5年前
- vignettesupload logo with transparent background3年前
- .RbuildignoreInitial commit5年前
- .gitignoreadded support to CRCh 38 (i.e., hg38-NCBI)3年前
- DESCRIPTIONUpdate DESCRIPTION1年前
- LICENSE.mdInitial commit5年前
- NAMESPACEupdated some functions and fixed unknown contig issue in readBam() when reading in a bam file aligned with different versions of hg19 or hg381年前
- NEWS.mdupdate news files3年前
- README.mdremove the hits badge7个月前
邀请码
版权所有:中国计算机学会技术支持:开源发展技术委员会
京ICP备13000930号-9
京公网安备 11010802047560号
cfDNAPro
Official tutorials
This landing page aims to provide a quick start. For in-depth documentation, please visit: https://cfdnapro.readthedocs.io/en/latest/
Declaration
cfDNAPro is designed for research only.
Why cfDNAPro?
Unlike genomic DNA, cfDNA has specific fragmentation patterns. The ambiguous definition of “fragment length” by various alignment software is raising concerns: see page 9 footnote in SAM file format spec: https://samtools.github.io/hts-specs/SAMv1.pdf\ cell-free DNA data fragmentomic analysis requires single-molecule level resolution, emphasising the importance of accurate/unbiased feature extraction. The traditional tools built for solid tissue sequencing do not consider the specific properties of cfDNA sequencing data (e.g., cfDNAs are naturally fragmented with a modal fragment size of 167bp, and di-/tri-nucleotide peaks in the length distributions). Researchers might inadvertently extract the features using a sub-optimised method.
cfDNAProis designed to resolve this issue and standardize the cfDNA fragmentomic analysis, complying with the existing building blocks in the bioconductor R ecosystem. We wish cfDNAPro to provide a catalyst for further improvements in the implementation and development of cfDNA biomarkers and multi-modal AI for various health conditions.Input
A paired-end sequencing bam file, with duplicates marked. (e.g., using the MarkDuplicates function from Picard).
Please do not impose any filtering on the bam files; For example, do not filter by the proper-pairs flag.
cfDNAProfilters the reads by following default criteria (You can toggle those criteria using parameters built-inreadBam()function):(1) Reads mapping qualities less than 30 were discarded;
(2) Reads must be paired. Of note, by default, cfDNAPro doesn’t impose filtration by “proper pair”;
(3) No duplicate;
(4) No secondary alignment;
(5) No supplementary alignment;
(6) No unmapped reads.
Note: remember to choose the correct
genome_label, a parameter inreadBam()function, based on the ref genome you used for alignment. At the moment, it supports three different ref genomes, hg19, hg38 and hg38-NCBI, For details see readBam() R documentation by typing?readBamin the R console or see source code:https://github.com/hw538/cfDNAPro/blob/master/R/readBam.ROutput
cfDNAProcan extract (i.e., “quantify in a standandised and robust way”) these features/bio-markers: - fragment length - fragment start/end/upstream/downstream motifs - copy number variation - single nucleotide mutation - more…Feature extraction depends on essential data objects/R packages in the Bioconductor ecosystem, such as
Rsamtools,plyranges,GenomicAlignments,GenomeInfoDbandBiostrings.Data engineering depends on packges in the tidyverse ecosystem, such as
dplyr, andstringr.All plots depend on
ggplot2R packge.For issues/inquiries, please contact:
Generic enquiry: Nitzan Rosenfeld Lab admin mailbox: bci-nrlab-admin@qmul.ac.uk
Fragment length, motif and CNV related questions: Haichao Wang: wanghaichao2014@gmail.com
SNV/SNP related questions: Paulius D. Mennea: paulius.mennea@cruk.cam.ac.uk
Installation
Option 1 (recommended): Use Docker or Singularity:
Thanks zetian-jia for building the docker image,
please refer to github.com/zetian-jia/cfDNAPro_docker
Docker
Singularity
Option 2: Use anaconda to build an env using the following codes:
Quick Start 1
Read bam file, return the fragment name (i.e. read name in bam file) and alignment coordinates in GRanges object in R. If needed, you can convert the GRanges into a dataframe and the fragment length is stored in the “width” column.
A screenshot of the output:
Quick Start 2
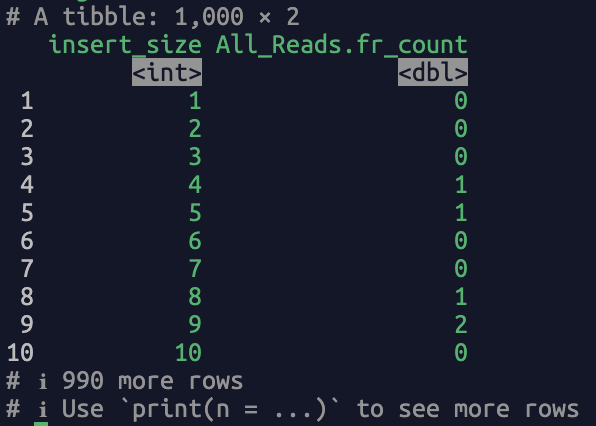
Read in bam file, return the fragment length counts. A straightforward and frequent user case: calculate the fragment size of a bam file, use the following code:
The returned dataframe contains two columns, i.e., “insert_size” (fragment length) and “All_Reads.fr_count” (the count of the fragment length). A screenshot of the output:
News
cfDNAPro paper is published on Genome Biology (May 2025)!
read_bam_insert_metricsfunction in cfDNAPro package. It is a tsv file containing two columns, i.e., “insert_size” (fragment length) and “All_Reads.fr_count” (the count of the fragment length). ### cfDNAPro 1.5.3 (Oct 2022)Citation
Please cite this paper:
Wang, H., Mennea, P.D., Chan, Y.K.E., Cheng, Z. et al. A standardized framework for robust fragmentomic feature extraction from cell-free DNA sequencing data. Genome Biol 26, 141 (2025). https://doi.org/10.1186/s13059-025-03607-5